Group 1
Nothing lasts forever, not even your problems. Stay Positive !
Project Objective:
Find the orthologs among the 5 species of beetles and annotate the orthologs.
Project Workflow:
Step 1: Construct a de novo transcriptome assembly.
a. Run Trinity to construct a primary assembly.
b. Run BUSCO to check the quality of the assembly. Use BUSCO.
Step 2: Filter transcripts with low expression.
a. Quantify the expression for each gene. Use salmon in galaxy.
- You can import the following galaxy history provided here. - Galaxy History
- This history has all the reads and the four de novo trinity assembly required to run salmon.
- Salmon is available as a tool in Galaxy. - Salmon in Galaxy
- Here are answers to some of the questions about the input data in salmon.
- Is this library mate-paired? = Paired-end
- Relative orientation of reads within a pair = Mates are oriented towards each other(I = inward)
- Specify the strandedness of the reads
Not stranded (U) = Not Stranded (U)
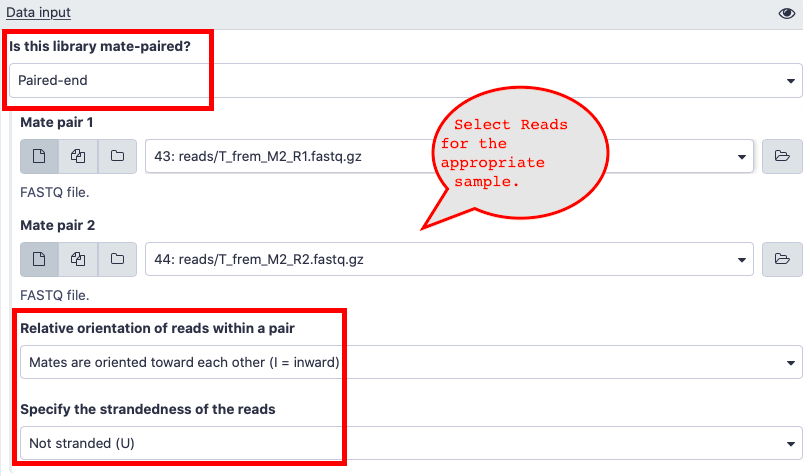
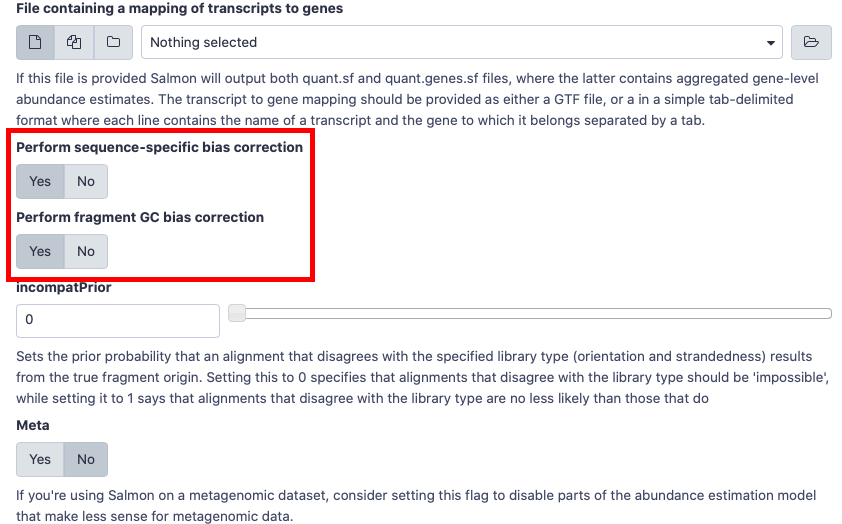
- Please choose
Yesfor gcBias and sequence-specific Bias when you run salmon.
b. Retain transcripts with a minimum of 5 TPM. Write python script.
c. Run BUSCO to check the quality of the filtered transcriptome. Use BUSCO.
Step 3: Identify the coding regions.
a. Using generated transcriptome from the previous step, run LongOrfs with threshold set to at least 100 aa length for each ORF. Use TransDecoder.LongOrfs
b. Using the predicted peptide sequences (.pep file) run BLASTP against Tribolium castaneum protein sequences. Use makeblastdb and blastp.
# Get the sequence file.
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/335/GCF_000002335.3_Tcas5.2/GCF_000002335.3_Tcas5.2_protein.faa.gz
# Uncompress the file.
gzip -d GCF_000002335.3_Tcas5.2_protein.faa.gz
# Make sure to create blastdb before this command.
# Example blastp.
blastp -query transdecoder_dir/longest_orfs.pep -db Tcas_protein_db.fasta -max_target_seqs 1 -outfmt 6 -evalue 1e-5 -num_threads 48 > blastp.outfmt6
c. Using the homology information from BLASTP, predict the coding sequence.
TransDecoder.Predict -t target_transcripts.fasta --retain_blastp_hits blastp.outfmt6
d. Run BUSCO to check the quality of the filtered transcriptome. Use BUSCO
Step 4: Find the Orthologs among 5 species.
a. Run all vs all BLAST among 5 species. Use makeblastdb and blastp
b. Pick the reciprocal Best BLAST hit (RBBH). Write a python script.
c. Run a 5 way script to pull out the orthologs among 5 species. Write a python script.
Step 5: Add annotation to the orthologs.
a. Using T_cas ids and T_cas reference gff files, add chromosome id (for example: LGX, LG2 etc.),chromosome name (Autosome/ Sex chromosome). Write python script.
# Get the gff file.
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/335/GCF_000002335.3_Tcas5.2/GCF_000002335.3_Tcas5.2_genomic.gff.gz
# Uncompress the file.
gzip -d GCF_000002335.3_Tcas5.2_genomic.gff.gz
Helpful Hints:
-
To use BUSCO, the first step is to get the lineage and then busco can be run as follows.
# Get the lineage wget https://busco.ezlab.org/datasets/endopterygota_odb9.tar.gz # Uncompress the directory. tar xvf endopterygota_odb9.tar.gz --gunzip # Activate busco environment conda activate busco # Run BUSCO run_busco --in transcriptome.fasta --out [output_directory_name] -l [path_to_]endopterygota_odb9 -m tran -c 48 - To use salmon, activate the conda environment and salmon can be run as follows.
# Activate salmon environment. conda activate salmon # Create an index for the transcriptome. salmon index -t transcriptome.fasta -i transcriptome_index # Quantify the expression. salmon quant -i transcriptome_index -l IU --seqBias --gcBias -1 Sample1_R1.fastq.gz -2 Sample1_R2.fastq.gz -p 48 --validateMappings -o Sample1 - TransDecoder has two programs. These two programs can be run as follows.
# To run LongOrfs with minimum ORF length as 100 amino acids. TransDecoder.LongOrfs -t target_transcripts.fasta -m 100 # To run Predict TransDecoder.Predict -t target_transcripts.fasta --retain_blastp_hits blastp.outfmt6
Reference:
De novo transcriptome assembly, functional annotation and differential gene expression analysis of juvenile and adult E. fetida, a model oligochaete used in ecotoxicological studies - PDF